[Courtesy: NatureNews]
Databases could soon be flooded with genome sequences from 25,000 tumours. Heidi Ledford looks at the obstacles researchers face as they search for meaning in the data.
When it was first discovered, in 2006, in a study of 35 colorectal cancers1, the mutation in the gene IDH1 seemed to have little consequence. It appeared in only one of the tumours sampled, and later analyses of some 300 more have revealed no additional mutations in the gene. The mutation changed only one letter of IDH1, which encodes isocitrate dehydrogenase, a lowly housekeeping enzyme involved in metabolism. And there were plenty of other mutations to study in the 13,000 genes sequenced from each sample. "Nobody would have expected IDH1 to be important in cancer," says Victor Velculescu, a researcher at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University in Baltimore, Maryland, who had contributed to the study.
But as efforts to sequence tumour DNA expanded, the IDH1 mutation surfaced again: in 12% of samples of a type of brain cancer called glioblastoma multiforme2, then in 8% of acute myeloid leukaemia samples3. Structural studies showed that the mutation changed the activity of isocitrate dehydrogenase, causing a cancer-promoting metabolite to accumulate in cells4. And at least one pharmaceutical company — Agios Pharmaceuticals in Cambridge, Massachusetts — is already hunting for a drug to stop the process.
Four years after the initial discovery, ask a researcher in the field why cancer genome projects are worthwhile, and many will probably bring up the IDH1 mutation, the inconspicuous needle pulled from a veritable haystack of cancer-associated mutations thanks to high-powered genome sequencing. In the past two years, labs around the world have teamed up to sequence the DNA from thousands of tumours along with healthy cells from the same individuals. Roughly 75 cancer genomes have been sequenced to some extent and published; researchers expect to have several hundred completed sequences by the end of the year.
The efforts are certainly creating bigger haystacks. Comparing the gene sequence of any tumour to that of a normal cell reveals dozens of single-letter changes, or point mutations, along with repeated, deleted, swapped or inverted sequences (see 'Genomes at a glance'). "The difficulty," says Bert Vogelstein, a cancer researcher at the Ludwig Center for Cancer Genetics and Therapeutics at Johns Hopkins, "is going to be figuring out how to use the information to help people rather than to just catalogue lots and lots of mutations". No matter how similar they might look clinically, most tumours seem to differ genetically. This stymies efforts to distinguish the mutations that cause and accelerate cancers — the drivers — from the accidental by-products of a cancer's growth and thwarted DNA-repair mechanisms — the passengers. Researchers can look for mutations that pop up again and again, or they can identify key pathways that are mutated at different points. But the projects are providing more questions than answers. "Once you take the few obvious mutations at the top of the list, how do you make sense of the rest of them?" asks Will Parsons, a paediatric oncologist at Baylor College of Medicine in Houston, Texas. "How do you decide which are worthy of follow up and functional analysis? That's going to be the hard part."
Because cancer is a disease so intimately associated with genetic mutation, many thought it would be amenable to genomic exploration through initiatives based on the collaborative model of the Human Genome Project. The International Cancer Genome Consortium (ICGC), formed in 2008, is coordinating efforts to sequence 500 tumours from each of 50 cancers. Together, these projects will cost in the order of US$1 billion. Eleven countries have already signed on to cover more than 20 cancers (see map). The ICGC includes two older, large-scale projects: the Cancer Genome Project, at the Wellcome Trust Sanger Institute near Cambridge, UK, and the US National Institutes of Health's Cancer Genome Atlas (TCGA). The Cancer Genome Project has churned out more than 100 partial genomes and roughly 15 whole genomes in various stages of completion, and intends to tackle 2,000–3,000 more over the next 5–7 years. TCGA, meanwhile, wrapped up a three-year, three-cancer pilot project last year, then launched a full-scale endeavour to sequence up to 500 tumours from each of more than 20 cancers over the next five years.
Although the groups collaborate, TCGA has not yet been able to fully join the ICGC owing to differences in privacy regulations governing access to genome data. For now, members of both consortia are sequencing a subset of tumour samples from each cancer type — around 100 — and will follow this by sequencing promising areas in the remaining 400. That's useful, says Joe Gray, a cancer researcher at Lawrence Berkeley National Laboratory in California, but it's just a start. "In the early days, I thought that doing a few hundred tumours would probably be sufficient," he says. "Even at the level of 1,000 samples, I think we're probably not going to have the statistics we want."
What bigger numbers could provide is more driver mutations like the one in IDH1. These could, researchers argue, provide the clearest route to developing new cancer therapies. Many scientists have looked for mutations that occur repeatedly in a given type of tumour. "If there are lots and lots of abnormalities of a particular gene, the most likely explanation is often that those mutations have been selected for by the cancers and therefore they are cancer-causing," says Michael Stratton, who co-directs the Cancer Genome Project. This approach has worked well in some cancers. For example, with a frequency of 12%, it is clear that the IDH1 mutation is a driver in glioblastoma. Such searches should be fruitful for cancers that have fewer mutations overall. The full genome sequence of acute myeloid leukaemia cells yielded just ten mutations in protein-coding genes, eight of which had not previously been linked with cancer5.
Other cancers have proved more challenging. IDH1 was overlooked at first, on the basis of the colorectal cancer data alone. It was not until the search was expanded to other cancers that its importance was revealed. Moreover, some mutations shown to be drivers haven't turned up as often as expected. "It's very clear, now that all the genes have been sequenced in this many tumours, you have drivers that are mutated at very low frequency, in less than 1% of the cancers," says Vogelstein. To find these low-frequency drivers, researchers are sampling heavily — sequencing 500 samples per cancer should reveal mutations that are present in as few as 3% of the tumours. Although they may not contribute to the majority of tumours, they may still have important biological lessons, says Stratton. "We need to know about these to understand the overall genomic landscape of cancer."
Another popular approach has been to look for mutations that cluster in a pathway, a group of genes that work together to carry out a specific process, even if the mutations strike it at different points. In an analysis of 24 pancreatic cancers6, for instance, Vogelstein and his colleagues identified 12 signalling pathways that had been altered. Nevertheless, Vogelstein cautions that this approach is not easy to pursue. Many pathways overlap, and their boundaries are unclear. And because many have been defined using data from different animals or cell types, they do not always match what's found in a specific human tissue. "When you layer on top of that the fact that the cancer cell is not wired the same as a normal cell, that raises even further difficulties," says Vogelstein.
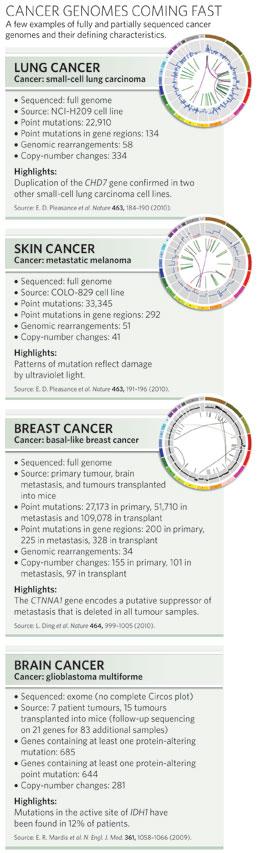
Separating drivers from passengers will become even more difficult as researchers move towards sequencing entire tumour genomes. To date, only a fraction of the existing cancer genomes are complete sequences. To keep costs low, most have covered only the exome, the 1.5% of the genome that directly codes for protein and is therefore the easiest to interpret. Assigning importance to a mutation found in the murky non-protein-coding depths of the genome will be more challenging, especially given that scientists don't yet know what function — if any — most of these regions usually serve. The vast majority of mutations fall here. The full genome sequence of a lung cancer cell line, for example, yielded 22,910 point mutations, only 134 of which were in protein-coding regions (see graphic, right)7. Nevertheless, finding them is worth the cost and effort, argues Stratton. "It could be that none of those mutations pertain to the causation of cancer," he says. "But it equally could be that some do. We'll never find out unless we systematically investigate."
Not everyone agrees. Some researchers argue that the costs of cancer-genome projects currently outweigh the benefits. Prices are poised to drop dramatically in the next few years as a new generation of sequencing machines comes online, says Ari Melnick, a cancer researcher at Weill Cornell Medical College in New York. "Why not wait for that?" he asks. In the meantime there are lower-hanging fruit to pick, says Stephen Elledge, a geneticist at Harvard Medical School in Boston, Massachusetts. Mutations that affect how many copies of a gene are found in a genome, he argues, are cheaper to assess and provide a more intuitive insight into biological processes. "If you delete something, you can turn a pathway off very efficiently," he says. "And if you amplify something, you can increase flow through the pathway. Making point mutations in genes to activate them is a little dicier."
Changes in gene copy number can be detected using fast, relatively inexpensive array-based technologies, but sequencing can provide a higher-resolution snapshot of these regions, says Elaine Mardis, a sequencing specialist at Washington University in St Louis, Missouri. Sequencing can enable researchers to map the boundaries of insertions and duplications with more precision and to catch tiny duplications or deletions that might have gone undetected by an array. Mardis, along with her colleague Richard Wilson and others, used sequencing to detect overlapping deletions in a breast cancer that had spread to other parts of the body (see page 999)8. The deletions spanned the region containing CTNNA1, a gene thought to suppress the spread, or metastasis, of cancer.
Meanwhile, cancer genomics is spreading out from under the large, centralized projects. For example, a $65-million, three-year paediatric-cancer genome project headed by researchers at St Jude Children's Research Hospital in Memphis, Tennessee, and Washington University aims to sequence 600 tumours. And more small projects seem poised to pop up. "Pretty much any cancer centre with any interest in the genomics of cancer is now buying these sequencers and using them," says Sam Aparicio, a cancer researcher at the University of British Columbia in Vancouver, Canada.
Part of the reason that cancer-genome proponents don't want to wait for sequencing costs to drop is that the real work starts after the sequencing is over. As Velculescu puts it, "Ultimately it's going to take good old-fashioned biology and experimental analyses to really determine what these mutations are doing." With this in mind, the US National Cancer Institute established two 2-year projects in September last year to develop high-throughput methods to test how the mutations identified by the TCGA pilot project affect cell function. The two centres — one at the Dana-Farber Cancer Center in Boston, and another at Cold Spring Harbor Laboratory in New York — aim to systematize the way that researchers pull other needles like the IDH1 mutation from the cancer-genomes haystack and make sense of them. The Boston team will systematically amplify and reduce the expression of genes of interest in cell cultures, and the Cold Spring Harbor centre will study cancer-associated mutations using tumours transplanted into mice.
In addition, large-scale projects are being run in parallel with the cancer-sequencing consortia to assess the effects of deleting each gene in the mouse genome, enabling researchers to learn more about the normal function of genes that are mutated in cancer. Sequencing is all very well, researchers have realized, but it won't be enough. "Some people say statistics should get us all the drivers that are worthwhile," says Lynda Chin, an investigator with TCGA at Harvard Medical School. "I don't agree with that. At the end of the day, we need these functional studies to prioritize the list of potential cancer-relevant candidates."

But as efforts to sequence tumour DNA expanded, the IDH1 mutation surfaced again: in 12% of samples of a type of brain cancer called glioblastoma multiforme2, then in 8% of acute myeloid leukaemia samples3. Structural studies showed that the mutation changed the activity of isocitrate dehydrogenase, causing a cancer-promoting metabolite to accumulate in cells4. And at least one pharmaceutical company — Agios Pharmaceuticals in Cambridge, Massachusetts — is already hunting for a drug to stop the process.
Four years after the initial discovery, ask a researcher in the field why cancer genome projects are worthwhile, and many will probably bring up the IDH1 mutation, the inconspicuous needle pulled from a veritable haystack of cancer-associated mutations thanks to high-powered genome sequencing. In the past two years, labs around the world have teamed up to sequence the DNA from thousands of tumours along with healthy cells from the same individuals. Roughly 75 cancer genomes have been sequenced to some extent and published; researchers expect to have several hundred completed sequences by the end of the year.
The efforts are certainly creating bigger haystacks. Comparing the gene sequence of any tumour to that of a normal cell reveals dozens of single-letter changes, or point mutations, along with repeated, deleted, swapped or inverted sequences (see 'Genomes at a glance'). "The difficulty," says Bert Vogelstein, a cancer researcher at the Ludwig Center for Cancer Genetics and Therapeutics at Johns Hopkins, "is going to be figuring out how to use the information to help people rather than to just catalogue lots and lots of mutations". No matter how similar they might look clinically, most tumours seem to differ genetically. This stymies efforts to distinguish the mutations that cause and accelerate cancers — the drivers — from the accidental by-products of a cancer's growth and thwarted DNA-repair mechanisms — the passengers. Researchers can look for mutations that pop up again and again, or they can identify key pathways that are mutated at different points. But the projects are providing more questions than answers. "Once you take the few obvious mutations at the top of the list, how do you make sense of the rest of them?" asks Will Parsons, a paediatric oncologist at Baylor College of Medicine in Houston, Texas. "How do you decide which are worthy of follow up and functional analysis? That's going to be the hard part."

Drivers wanted
Because cancer is a disease so intimately associated with genetic mutation, many thought it would be amenable to genomic exploration through initiatives based on the collaborative model of the Human Genome Project. The International Cancer Genome Consortium (ICGC), formed in 2008, is coordinating efforts to sequence 500 tumours from each of 50 cancers. Together, these projects will cost in the order of US$1 billion. Eleven countries have already signed on to cover more than 20 cancers (see map). The ICGC includes two older, large-scale projects: the Cancer Genome Project, at the Wellcome Trust Sanger Institute near Cambridge, UK, and the US National Institutes of Health's Cancer Genome Atlas (TCGA). The Cancer Genome Project has churned out more than 100 partial genomes and roughly 15 whole genomes in various stages of completion, and intends to tackle 2,000–3,000 more over the next 5–7 years. TCGA, meanwhile, wrapped up a three-year, three-cancer pilot project last year, then launched a full-scale endeavour to sequence up to 500 tumours from each of more than 20 cancers over the next five years.

Although the groups collaborate, TCGA has not yet been able to fully join the ICGC owing to differences in privacy regulations governing access to genome data. For now, members of both consortia are sequencing a subset of tumour samples from each cancer type — around 100 — and will follow this by sequencing promising areas in the remaining 400. That's useful, says Joe Gray, a cancer researcher at Lawrence Berkeley National Laboratory in California, but it's just a start. "In the early days, I thought that doing a few hundred tumours would probably be sufficient," he says. "Even at the level of 1,000 samples, I think we're probably not going to have the statistics we want."
“Even at the level of 1,000 samples, I think we're probably not going to have the statistics we want.”
Other cancers have proved more challenging. IDH1 was overlooked at first, on the basis of the colorectal cancer data alone. It was not until the search was expanded to other cancers that its importance was revealed. Moreover, some mutations shown to be drivers haven't turned up as often as expected. "It's very clear, now that all the genes have been sequenced in this many tumours, you have drivers that are mutated at very low frequency, in less than 1% of the cancers," says Vogelstein. To find these low-frequency drivers, researchers are sampling heavily — sequencing 500 samples per cancer should reveal mutations that are present in as few as 3% of the tumours. Although they may not contribute to the majority of tumours, they may still have important biological lessons, says Stratton. "We need to know about these to understand the overall genomic landscape of cancer."
Another popular approach has been to look for mutations that cluster in a pathway, a group of genes that work together to carry out a specific process, even if the mutations strike it at different points. In an analysis of 24 pancreatic cancers6, for instance, Vogelstein and his colleagues identified 12 signalling pathways that had been altered. Nevertheless, Vogelstein cautions that this approach is not easy to pursue. Many pathways overlap, and their boundaries are unclear. And because many have been defined using data from different animals or cell types, they do not always match what's found in a specific human tissue. "When you layer on top of that the fact that the cancer cell is not wired the same as a normal cell, that raises even further difficulties," says Vogelstein.
How much is enough?
Separating drivers from passengers will become even more difficult as researchers move towards sequencing entire tumour genomes. To date, only a fraction of the existing cancer genomes are complete sequences. To keep costs low, most have covered only the exome, the 1.5% of the genome that directly codes for protein and is therefore the easiest to interpret. Assigning importance to a mutation found in the murky non-protein-coding depths of the genome will be more challenging, especially given that scientists don't yet know what function — if any — most of these regions usually serve. The vast majority of mutations fall here. The full genome sequence of a lung cancer cell line, for example, yielded 22,910 point mutations, only 134 of which were in protein-coding regions (see graphic, right)7. Nevertheless, finding them is worth the cost and effort, argues Stratton. "It could be that none of those mutations pertain to the causation of cancer," he says. "But it equally could be that some do. We'll never find out unless we systematically investigate."
Not everyone agrees. Some researchers argue that the costs of cancer-genome projects currently outweigh the benefits. Prices are poised to drop dramatically in the next few years as a new generation of sequencing machines comes online, says Ari Melnick, a cancer researcher at Weill Cornell Medical College in New York. "Why not wait for that?" he asks. In the meantime there are lower-hanging fruit to pick, says Stephen Elledge, a geneticist at Harvard Medical School in Boston, Massachusetts. Mutations that affect how many copies of a gene are found in a genome, he argues, are cheaper to assess and provide a more intuitive insight into biological processes. "If you delete something, you can turn a pathway off very efficiently," he says. "And if you amplify something, you can increase flow through the pathway. Making point mutations in genes to activate them is a little dicier."
Changes in gene copy number can be detected using fast, relatively inexpensive array-based technologies, but sequencing can provide a higher-resolution snapshot of these regions, says Elaine Mardis, a sequencing specialist at Washington University in St Louis, Missouri. Sequencing can enable researchers to map the boundaries of insertions and duplications with more precision and to catch tiny duplications or deletions that might have gone undetected by an array. Mardis, along with her colleague Richard Wilson and others, used sequencing to detect overlapping deletions in a breast cancer that had spread to other parts of the body (see page 999)8. The deletions spanned the region containing CTNNA1, a gene thought to suppress the spread, or metastasis, of cancer.
Meanwhile, cancer genomics is spreading out from under the large, centralized projects. For example, a $65-million, three-year paediatric-cancer genome project headed by researchers at St Jude Children's Research Hospital in Memphis, Tennessee, and Washington University aims to sequence 600 tumours. And more small projects seem poised to pop up. "Pretty much any cancer centre with any interest in the genomics of cancer is now buying these sequencers and using them," says Sam Aparicio, a cancer researcher at the University of British Columbia in Vancouver, Canada.
ADVERTISEMENT

Part of the reason that cancer-genome proponents don't want to wait for sequencing costs to drop is that the real work starts after the sequencing is over. As Velculescu puts it, "Ultimately it's going to take good old-fashioned biology and experimental analyses to really determine what these mutations are doing." With this in mind, the US National Cancer Institute established two 2-year projects in September last year to develop high-throughput methods to test how the mutations identified by the TCGA pilot project affect cell function. The two centres — one at the Dana-Farber Cancer Center in Boston, and another at Cold Spring Harbor Laboratory in New York — aim to systematize the way that researchers pull other needles like the IDH1 mutation from the cancer-genomes haystack and make sense of them. The Boston team will systematically amplify and reduce the expression of genes of interest in cell cultures, and the Cold Spring Harbor centre will study cancer-associated mutations using tumours transplanted into mice.
“It's going to take good old-fashioned biology to really determine what these mutations are doing.”
See also News and Views, page 989.
-
References
- Sjöblom, T. et al. Science 314, 268-274 (2006). | Article | PubMed | ISI | ChemPort |
- Parsons, D. W. et al. Science 321, 1807-1812 (2008). | Article | PubMed | ChemPort |
- Mardis, E. R. et al. N. Engl. J. Med. 361, 1058-1066 (2009). | Article | PubMed | ChemPort |
- Dang, L. et al. Nature 462, 739-744 (2009). | Article | PubMed | ChemPort |
- Ley, T. J. et al. Nature 456, 66-72 (2008). | Article | PubMed | ChemPort |
- Jones, S. et al. Science 321, 1801-1806 (2008). | Article | PubMed | ChemPort |
- Pleasance, E. D. et al. Nature 463, 184-190 (2010). | Article | PubMed | ChemPort |
- Ding, L. et al. Nature 464, 999-1005 (2010). | Article

1 comment:
Hello,
I have a question about your blog. Please email me!
Thanks,
David
Post a Comment